Japanese
English
- 有料閲覧
- Abstract 文献概要
- 1ページ目 Look Inside
緒言 1876年Thomsen1)が特異な筋強直性反應を呈する一つの遺傳性疾患として先天性筋強直症(M-yotonia congenita)〔別名「トムゼン」氏病〕を記載したが,その後1896年Jolley1)がこの疾患に筋強直性反應の外に筋榮養障碍を伴う型のあることを指摘した。然し尚之は「トムゼン」氏病の非完型の疾患と見做されていた。然るに1909年Steinert3)が,又米國では同年Batten及びGi-bbs4)が各々獨立して筋榮養障碍を伴う型のものを綜合的に検索して,之等の例では筋強直性反應はその分布が限局していて,筋榮養障碍が特徴的な症状であつて「トムゼン」氏病が筋榮養障碍を何れにも見られないのと對比的であることを重視して,之は「トムゼン」氏病とは獨立した疾患として取扱うべきであるとしてDystrophia myo-tonica (Myotonia atrophica)〔筋強直性 「デイストロフイー」症〕と記載した。續いて1912年Curschmann5)は筋榮養障碍の外に白内障,禿頭,睾丸萎縮その他の内分泌系臟器機能の變調等の徴候が合併していて之が筋強直性「デイストロフイー」症の極めて特異的な症候群であることを強調した。
本邦では1916年横森6)の報告が最初でその後阿部7),廣田8),安生9),和田10),前田11),柴12),熊谷13),石川,武市14),柴田15),俵16),猪瀨17),吉田18),冲中・栗本19),大屋20),市岡21),太田22),井上23),柴田24)の18家族,23例の報告がある。之等の報告では,東京,東北,新潟,名古屋,岡山,京都に分布を見るがその他の府縣からの報告はない。
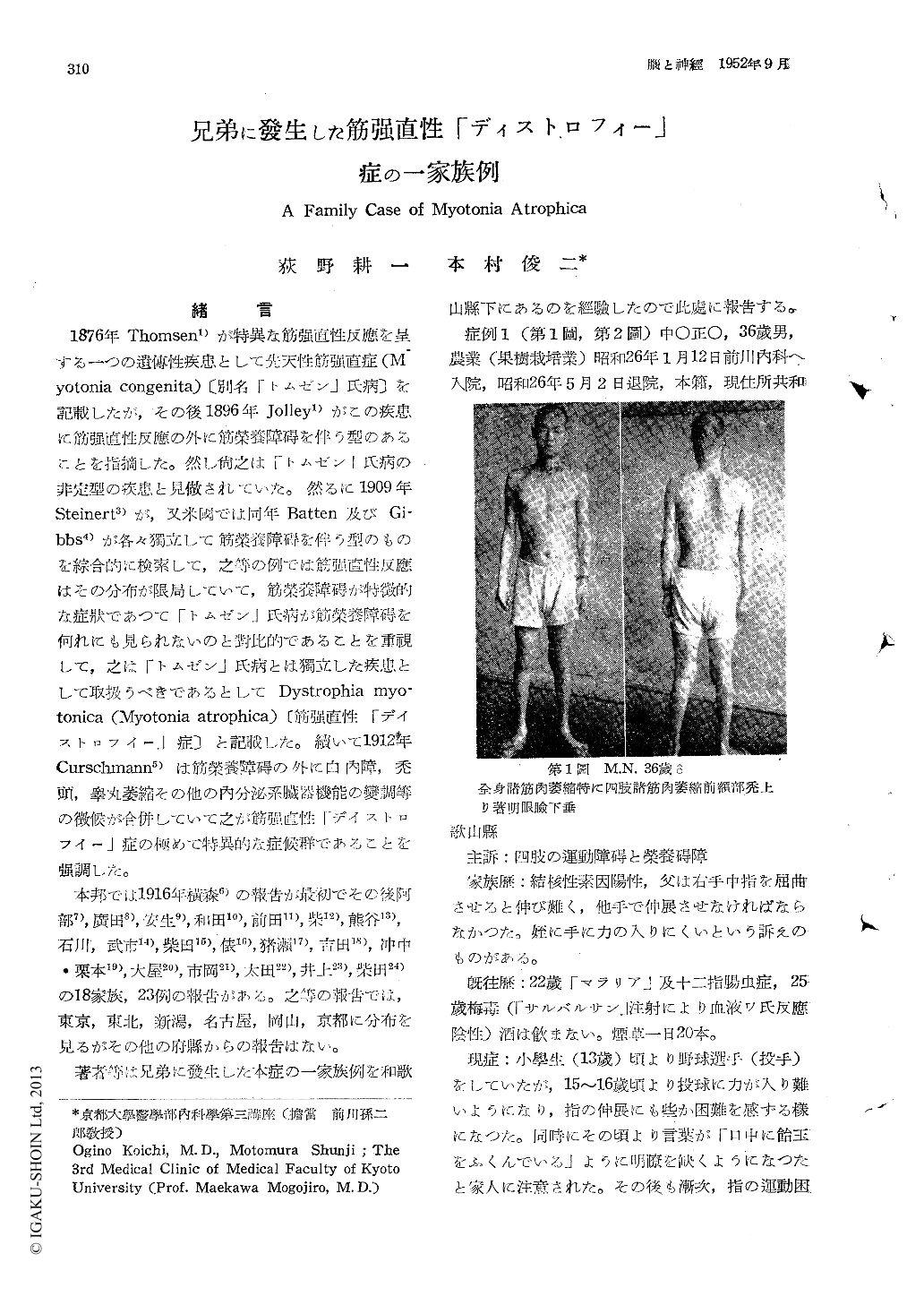
There has been reported in Japan, 18 family cases (23 indivifual cases) of the myotonia atrophica (Dystrophia myotonica), in the regions of Sendai, Tokyo, Niigata, Okayama, Kyoto, Nagoya, but not in other regions. Recently we experienced a family case of this rare disease, the symptoms of which manifested themselves in two brothers, in the Wakayama Prefecture as follows. The elder one was a farmer aged 36. He already began to feel the difficulty to relax promptly his fingers after a forceful grasp at the age of 15. Thereafter, this disorder gradually intensified, and, besides this 2 years ago, the trophic disturbance appeared in his extremities, so that he became hard to walk, especially to go up the steps. Moreover, he recently was suffering from the impotency. The patient showed the delayed relaxation of muscles not only after the tightly grasp (active myotonic reaction), but also by the mechanical or electrical stimulations (passive myotonic reac-tion). The muscles in his extremities, face and neck was so markedly atrophic that he showed the myasthenic facies and the poor en-unciation. Besides, we found the testicular atrophy, the incipient cataract, the baldness of his fore-head and the increase of creatine in urine. The younger one was a techncian aged 32 in a wire factory. He first noticed 2 years ago, the delay in relaxing his grasp at opening the door, and then this disorder became more heavily. He also showed the active and passive myotonic reaction, the slight muscular atrophy in the nape of the neck and the sternocleido-mastoids, the baldness of his fore-head and the increase of creatine in urine, but not the cataract nor the testicular atrophy. Nowadays, most students support the opinion of Steinert that this disease is quite different from the myotonia congenita but a part of students are still of opinion that this is an atypical form of the myotonia congenita. According to our family case, we also support the Steinert's opinion, because the younger brother showed the slight muscular atrophy in some part in spite of the incipient state of the disease. And we believe that the most characteristic and important symptom of this disease in not a myotonia but a muscular dystrophy. Therefore, for the treat-ment of the patient with the marked muscular dystrophy, it should not be recommandable to administer for long duration the quinine, for this drug is originally harmful to the muscular protoplasm and has many bothersome side-effects. We tried to the elder patient the glycocoll (10gr/day) for long duration as in the case of the progressive muscular dystrophy. Thereafter about 45 days, his condition so much improved, but not perfectly, that his grasping power increased, the walking became easily, the creatine in urine decreased, but, in spite of these improvement, the myotonia did'nt become worse but slightly well.
Copyright © 1952, Igaku-Shoin Ltd. All rights reserved.

