Japanese
English
- 有料閲覧
- Abstract 文献概要
- 1ページ目 Look Inside
I.はじめに
“モヤモヤ”病の家族内発生の報告は散見されるが,1家系中2例が大部分であり,その頻度は7-10%といわれている8).その発症については遺伝的素因の関与が考えられているが,いまだ明らかではない.われわれは同胞および母子の3人に発生した“モヤモヤ”病の1家系を経験した.これらの遺伝的な関与を示唆する症例について,若干の文献的考察を加えて報告する.
Abstract
One familial case of “moyamoya” disease affecting three patients is reported.
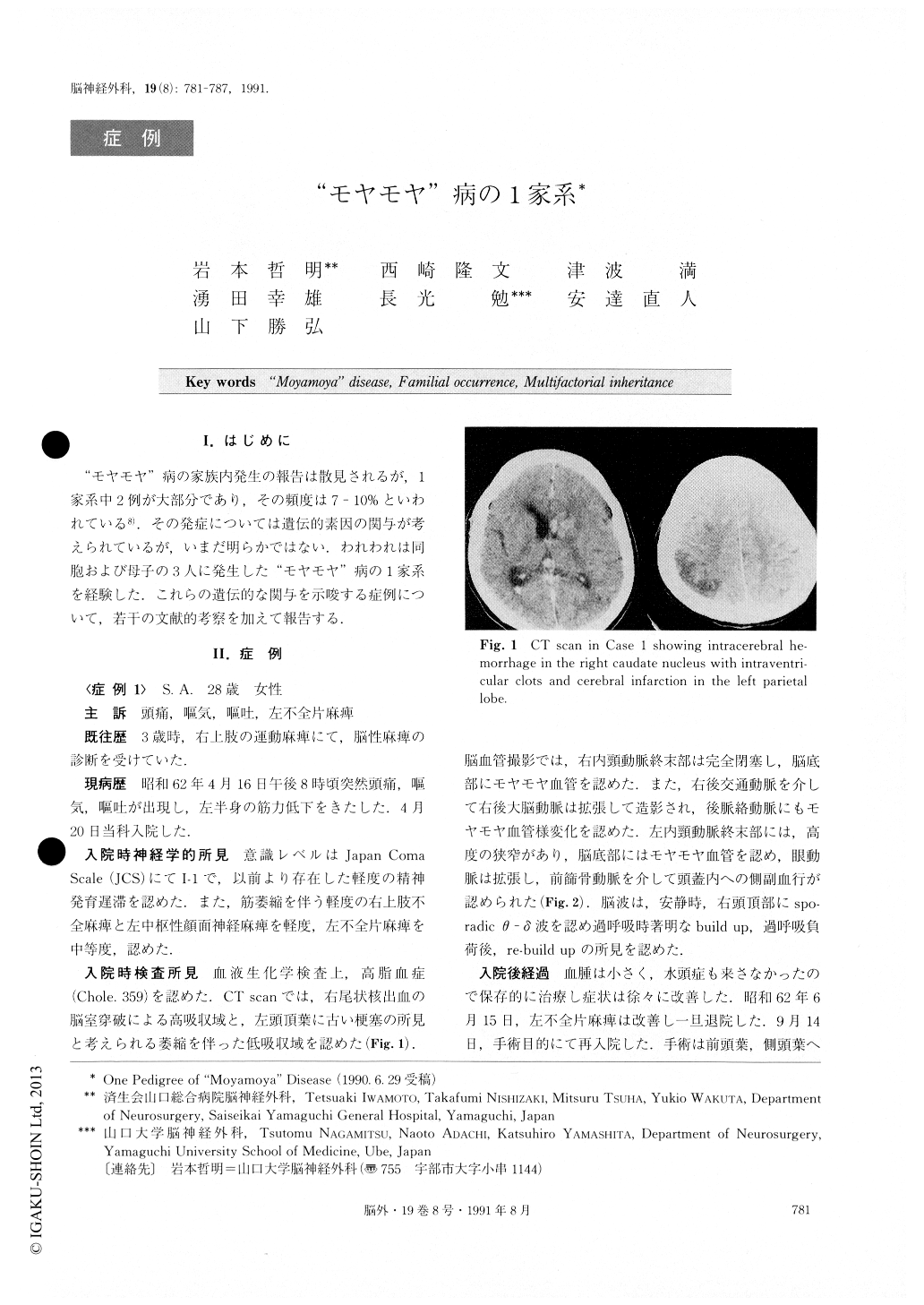
The patient in Case 1 was a 28-year-old female. She had suffered from motor weakness of the right limbs in her infantile period. She visited our hospital because of sudden headache and left motor weakness associated with nausea and vomiting. On admission, CT scan re-vealed cerebral hemorrhage in the right caudate nuc-leus with intraventricular clots and infarction in the left parietal lobe. Angiography showed stenosis of the left ICA terminal portion and occlusion of the right side, with moyamoya vessels in the basal area. of moya-moya disease ; report of three Japanese families. J Neurosurg 42 : 208-214, 1979 The patient in Case 2 was a 54-year-old female, who was the mother of Case 1. After an operation for acute upper intestinal bleeding, she suffered from cerebral in-farction. CT scan revealed large low density areas in the territory of the bilateral MCA. Angiography showed stenosis of the bilateral ICA terminal portions, occlusion of the right MCA, stenosis of the left MCA, and moyamoya vessels in the basal area.
The patient in Case 3 was a 40-year-old female, who was a younger sister of Case 2. She had a convulsive attack in her infantile period. She visited our hospital because of gradually worsening headache. CT scan re-vealed multiple infarctions in the left paraventricle, the right parieto-occipital and occipital lobe. Angiography showed occlusion of the bilateral ICA terminals with moyamoya vessels in the basal and the ethmoidal areas. The patient in Case 2 died immediately. Surgery for reconstruction of hemodynamics was performed in Case 1 and 3.
The rate of the familial occurrence of “moyamoya” disease is said to range from 7% to 10%. However, most of these cases have involved only two patients, and thus our case is rare (only four similar cases have been reported previously) . In general, it is considered that the mode of inheritance of “moyamoya” disease is mul-tifactorial inheritance, and such cases strongly suggest involvement to a genetic factor. More thorough screen-ing tests would reveal a higher rate of familial occur-rence, and consequently, facilitate early diagnosis and treatment.
Copyright © 1991, Igaku-Shoin Ltd. All rights reserved.

